摘 要:開(kāi)發(fā)新型純碳水化合物燃料作為新能源時(shí),必須預(yù)測(cè)和篩選純碳水化合物的物理性質(zhì),從而找到可能合適的化學(xué)物質(zhì),然而由實(shí)驗(yàn)來(lái)逐一確定大量分子的物理性質(zhì)既耗時(shí)又昂貴。研究發(fā)現(xiàn),運(yùn)用人工智能網(wǎng)絡(luò) 定量構(gòu)效關(guān)系(ANN—QSPR)算法來(lái)建立純碳水化合物物理性質(zhì)的計(jì)算模型可以起到事半功倍的效果。基于DIPPR 801數(shù)據(jù)庫(kù)中的純組分性質(zhì)和DragonX軟件包計(jì)算了相應(yīng)碳水化合物的分子描述符,所構(gòu)建的模型結(jié)合了定量構(gòu)效關(guān)系(QSPR)和兩層前饋人工智能網(wǎng)絡(luò)(ANN)。由此建立了多個(gè)全面而可靠的模型來(lái)預(yù)測(cè)新型純碳水化合物燃料的各種物理性質(zhì),包括正常沸點(diǎn)、閃點(diǎn)、燃燒焓、蒸發(fā)焓、液體密度、表面張力、液體的黏度和熔點(diǎn)等。為了提高模型中數(shù)據(jù)集之間的一致性,還引入了主成分分析法(PCA),以進(jìn)一步消除分子描述符值的維數(shù)。另外,通過(guò)共識(shí)建模進(jìn)行交叉驗(yàn)證,減少了不確定性的影響,提高了模型的預(yù)測(cè)精度。
關(guān)鍵詞:人工智能網(wǎng)絡(luò) 定量構(gòu)效關(guān)系 新型純碳水化合物燃料 新能源 物理性質(zhì) 預(yù)測(cè) 模型構(gòu)建
ANN-QSPR models for the predication of physical properties of a new-type carbohydrate fuel
Abstract:When a pure carbohydrate compound is developed as a novel combustion fuel,its physical properties of each component will be first necessarily predicted and screened.However,the experimental determination of these properties for a huge amount of molecules can be very time consuming and costly.In view of this,the artificial neural network-quantitative structure property relationships (ANN—QSPR) algorithm was applied to build the desired models.Molecular descriptors were calculated based on a large number of pure components with evaluated values in DIPPR 801 database and the software package DragonX.The models developed were combinations of QSPR and two layer feed forward ANN:Thus the relatively comprehensive and reliable models were developed for predicting physical properties,including normal boiling point,flash point,enthalpy of combustion,enthalpy of vaporization,liquid density,surface tension,liquid viscosity,melting point,etc.For improving the consistency,principal component analysis(PCA)was introduced to further eliminate the dimensions of molecular descriptor values.Finally,the idea of cross-validation for consensus modeling is further utilized to improve the predictive quality of obtained models.
Key words:ANN(artificial neural network),QSPR(qualitative structure property relationships),QSAR(qualitative structure activity relationships),new-type carbohydrate fuel,new energy source,physical property,forecast,modeling
化石燃料的不可再生性,迫使人們不斷研發(fā)新能源,以滿足社會(huì)存在和發(fā)展的需要。過(guò)去,當(dāng)研究人員開(kāi)發(fā)新型燃料時(shí),首要考慮的問(wèn)題是純碳水化合物的物理性質(zhì),其中包括了標(biāo)準(zhǔn)沸點(diǎn)、閃點(diǎn)、燃燒焓、蒸發(fā)焓、液體密度、表面張力、液體黏度和熔點(diǎn)。然而通過(guò)實(shí)驗(yàn)來(lái)逐一確定大量分子的性質(zhì)非常耗時(shí)和昂貴[1],所以人們迫切希望能構(gòu)建各種模型來(lái)對(duì)大量分子進(jìn)行篩選和預(yù)測(cè),從而找到可能合適的化學(xué)物質(zhì)。
基于上述原因,從現(xiàn)有文獻(xiàn)中可以發(fā)現(xiàn)人們已發(fā)展了各種用于預(yù)測(cè)物理性質(zhì)的方法。不過(guò),包括量子力學(xué)或詳細(xì)動(dòng)力機(jī)理在內(nèi)的那些尖端、高級(jí)的性質(zhì)研究方法同樣很耗時(shí)間,因此不適合用于篩選工作。目前使用最為廣泛的方法可以分為以下兩大類:
1)第一類方法的依據(jù)是基團(tuán)貢獻(xiàn)(OC)算法,其基本概念是決定物質(zhì)性質(zhì)常數(shù)的分子間作用力通常都取決于各分子的原子之間的鍵[2]。時(shí)至今日,人們已發(fā)展出了許多基于GC的方法。不過(guò)GC法也有一些重大缺陷,那就是無(wú)法獲得立體異構(gòu)體的確鑿結(jié)果[3],其所得結(jié)果通常也不是很精確[1-2]。
2)在最近幾年,人們采用了另一類被稱作定量結(jié)構(gòu)性質(zhì)關(guān)系(QSPR) [4]的方法米克服上述缺陷[5-6],此類關(guān)系有時(shí)也被稱作定量結(jié)構(gòu)一活性關(guān)系(QSAR)。QSPR的基本假設(shè)是:結(jié)構(gòu)相似的對(duì)象會(huì)展現(xiàn)相似的性質(zhì),因此可用數(shù)據(jù)分析法和統(tǒng)計(jì)法對(duì)此進(jìn)行大致的描述,從而構(gòu)建出各種模型;根據(jù)從結(jié)構(gòu)或拓?fù)渲笖?shù)到電子或量子化學(xué)性質(zhì)的各種參數(shù)[5-6] (這些參數(shù)通常被稱作分子描述符[7],可根據(jù)維數(shù)將其分為不同的類別[3]),可以準(zhǔn)確地通過(guò)這些模型預(yù)測(cè)化合物的生物活性或性質(zhì)。人們通常會(huì)在QSPR算法中采用某些多變量分析工具,諸如偏最小二乘法或PLSL[8]等。
最近,一種新方法——人工智能網(wǎng)絡(luò)(ANN)又被引入這一領(lǐng)域,并迅速成為研究結(jié)構(gòu)—性質(zhì)和結(jié)構(gòu)活性相互關(guān)系[9]的方法之一。因此,我們將在此項(xiàng)研究中展示一種基于人工智能網(wǎng)絡(luò)定量結(jié)構(gòu)性質(zhì)關(guān)系法(ANN-QSPR)的新方法,該方法用于篩選和預(yù)測(cè)純碳水化合物的性質(zhì)可以起到事半功倍的效果,從而有助于新型燃料的開(kāi)發(fā)。
1 材料和方法
1.1 材料
在構(gòu)建用于預(yù)測(cè)物理性質(zhì)的模型時(shí),所采用數(shù)據(jù)集的質(zhì)量和全面性將對(duì)其準(zhǔn)確性和可靠性產(chǎn)生很大影響,特別是對(duì)于那些需處理大量實(shí)驗(yàn)數(shù)據(jù)的模型[10]。在本次研究中,由于DIPPR 801[11]數(shù)據(jù)庫(kù)中含有許多純組分的性質(zhì),因此筆者采用了這一數(shù)據(jù)庫(kù)進(jìn)行計(jì)算和建模,并用軟件包DragonX[12]計(jì)算了相應(yīng)碳水化合物的分子描述符。考慮到當(dāng)前研究中會(huì)產(chǎn)生大量的分子結(jié)構(gòu),我們?cè)诮V屑{入了900個(gè)分子描述符,其中包括所有的零維、一維和二維描述符[12]。
1.2 數(shù)據(jù)預(yù)處理
在QSPR建模中,結(jié)構(gòu)異常值是影響模型精度的主要因素,所以在將數(shù)據(jù)庫(kù)用于模型構(gòu)建前,要先通過(guò)主成分分析法(PCA) [3,8]將結(jié)構(gòu)不同的化合物排除在外,同時(shí)也要排除非碳水化合物。最終,分別研究了純碳水化合物915、507、940、467、693、544、462和915在以下方面的相關(guān)數(shù)值:標(biāo)準(zhǔn)沸點(diǎn)、閃點(diǎn)、燃燒焓、蒸發(fā)焓、液體密度、表面張力、液體黏度和熔點(diǎn)。最初筆者展示了900個(gè)分子描述符,這一數(shù)目超過(guò)或大致等于所研究純碳水化合物的數(shù)目。ANN為非線性關(guān)系的結(jié)構(gòu),因此所提供的分子描述符數(shù)目將大大超過(guò)合適的數(shù)目,并對(duì)模型造成不利影響。在此項(xiàng)研究中,我們使用了遞歸共線診斷(SCD)程序來(lái)降低維數(shù)和去除多余的描述符[8,13]。此外,還排除了對(duì)所有純碳水化合物而言皆為常數(shù)值的分子描述符。用于建模的分子描述符的相應(yīng)數(shù)日為l71、153、172、152、l60、163和170。
1.3 模型構(gòu)建
在對(duì)數(shù)據(jù)進(jìn)行預(yù)處理后,下一個(gè)計(jì)算步驟——也可能是最重要的一個(gè)步驟——是找出分子描述符和碳水化合物物理性質(zhì)之間的關(guān)系。因此筆者采用了人工神經(jīng)網(wǎng)絡(luò)的非線性數(shù)學(xué)方法。
人工神經(jīng)網(wǎng)絡(luò)被廣泛用于許多科學(xué)和工程應(yīng)用領(lǐng)域,例如計(jì)算不同純化合物的物理和化學(xué)性質(zhì)[14]。讀者可在其他文獻(xiàn)中找到ANN的工作原理說(shuō)明[14-15]。在此項(xiàng)研究中,采用了MATLAB軟件來(lái)構(gòu)建ANN-QSPR模型。通過(guò)ANN工具箱,構(gòu)建了用于建模的兩層前饋ANN。圖l中顯示了兩層前饋ANN的結(jié)構(gòu)。
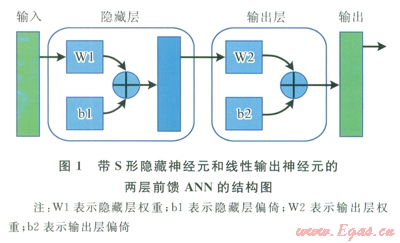
首先,在[-1,1]的范圍內(nèi)對(duì)純碳水化合物的所有性質(zhì)值進(jìn)行正交化,以便降低計(jì)算誤差,尤其是截?cái)嗾`差。然后用相同的方法對(duì)所有的分子描述符進(jìn)行標(biāo)準(zhǔn)化。這一正交化過(guò)程的具體操作是將性質(zhì)參數(shù)或描述符的最小值和最大值分別設(shè)為-l和1,然后相應(yīng)地?cái)M合其他數(shù)值。
其次,將數(shù)據(jù)庫(kù)分為3個(gè)子類,分別為“訓(xùn)練”集、“驗(yàn)證”集和“測(cè)試”集。“訓(xùn)練”集用于生成主方案或人工神經(jīng)網(wǎng)絡(luò)的結(jié)構(gòu)。“驗(yàn)證”集用于檢測(cè)訓(xùn)練終點(diǎn),也就是說(shuō),如果最新的受訓(xùn)模型正開(kāi)始使“驗(yàn)證”集中的預(yù)測(cè)數(shù)值變差,則應(yīng)停止訓(xùn)練過(guò)程。“測(cè)試”集被認(rèn)為是所獲模型在預(yù)測(cè)能力和質(zhì)量方面的一個(gè)重要指征。應(yīng)隨機(jī)開(kāi)展3個(gè)子類的數(shù)據(jù)選擇過(guò)程。在此項(xiàng)研究中,“訓(xùn)練”“驗(yàn)證”和“測(cè)試”集分別占80%、l0%、10%。從其他各類文獻(xiàn)中可以發(fā)現(xiàn)[15-16],主數(shù)據(jù)庫(kù)3個(gè)子類所分配的百分比將影響到模型的準(zhǔn)確性。
接下來(lái)需要生成ANN模型。事實(shí)上,這一生成過(guò)程其實(shí)是一個(gè)確定加權(quán)矩陣和偏倚向量的過(guò)程[15,17],應(yīng)通過(guò)目標(biāo)函數(shù)的最小化來(lái)獲取這些參數(shù)[1,10,14]。筆者采用了列文伯格—馬夸爾特(Levenberg-Marquardt,LM)反向傳播算法來(lái)實(shí)現(xiàn)目標(biāo)參數(shù)的最小化[17]。
2 結(jié)果與討論
按照上述步驟,獲得了各個(gè)兩層前饋神經(jīng)網(wǎng)絡(luò),以用于預(yù)測(cè)純碳水化合物的標(biāo)準(zhǔn)沸點(diǎn)、閃點(diǎn)、燃燒焓、蒸發(fā)焓、液體密度、表面張力、液體黏度和熔點(diǎn)。也有其他更為準(zhǔn)確的最小化方法,但它們需要的收斂時(shí)間要長(zhǎng)得多。LM反向傳播法是訓(xùn)練人工神經(jīng)網(wǎng)絡(luò)時(shí)最為常用的算法[18]。
最后應(yīng)固定神經(jīng)元的數(shù)目。這一數(shù)目取決于測(cè)試和嘗試結(jié)果;神經(jīng)元的最佳數(shù)目通常介于10~20[1,10,14],筆者將神經(jīng)元數(shù)目固定為15。剩下的工作則是生成一個(gè)ANN模型。
圖2~9中顯示了所構(gòu)建模型預(yù)測(cè)結(jié)果與實(shí)驗(yàn)數(shù)據(jù)之間的比較。表l中顯示了每個(gè)模型的平均相對(duì)誤差和最大相對(duì)誤差。
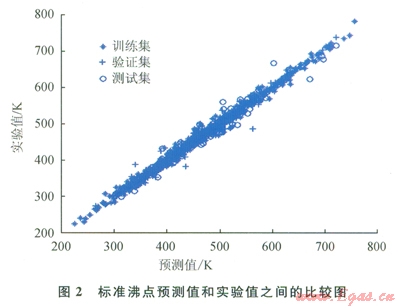
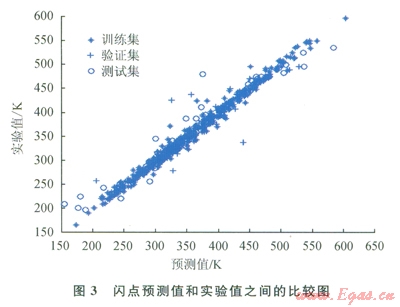
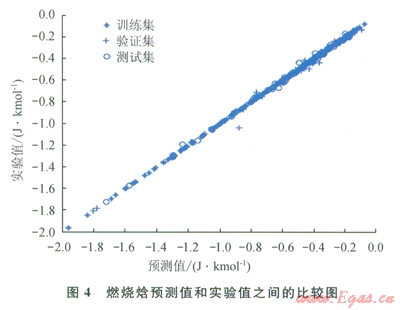
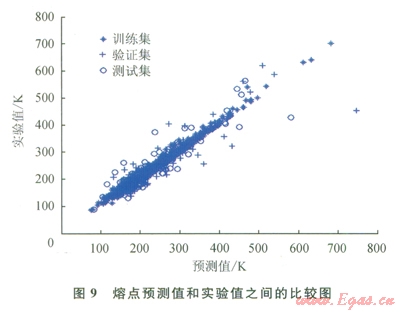
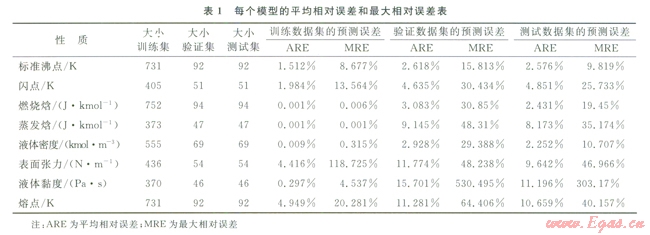
仔細(xì)研究所獲結(jié)果,可以發(fā)現(xiàn)“訓(xùn)練”集的相對(duì)誤差總是小于“驗(yàn)證”集或“測(cè)試”集的相對(duì)誤差,這主要是因?yàn)?ldquo;驗(yàn)證”集或“測(cè)試”集對(duì)ANN的訓(xùn)練方式?jīng)]有直接影響。因此,對(duì)人工神經(jīng)網(wǎng)絡(luò)進(jìn)行了修正,使其更適合于“訓(xùn)練”集的數(shù)據(jù)。可以把訓(xùn)練過(guò)程想象為找出方程組的待定系數(shù)。
化合物的數(shù)目表明了方程式數(shù)目,而分子描述符的數(shù)目則表明了系數(shù)數(shù)目。因此,化合物數(shù)目超出分子描述符數(shù)目的比率越小,ANN就能在“訓(xùn)練”集中表現(xiàn)得越好。不過(guò),對(duì)不在“訓(xùn)練”集內(nèi)的化合物來(lái)說(shuō),以這種方法構(gòu)建的ANN無(wú)法保證其預(yù)測(cè)水平。舉例來(lái)說(shuō)(如表l所示),對(duì)于所構(gòu)建的蒸發(fā)焓或燃燒焓模型而言,“測(cè)試”集的平均相對(duì)誤差遠(yuǎn)遠(yuǎn)大于“訓(xùn)練”集的平均相對(duì)誤差。解決此問(wèn)題的一個(gè)可能方案是采用PCA[8]:用PCA來(lái)處理描述符,然后找到得分,取前30列的得分(含上述信息99%的內(nèi)容),最后按照上述步驟構(gòu)建一個(gè)新的ANN。
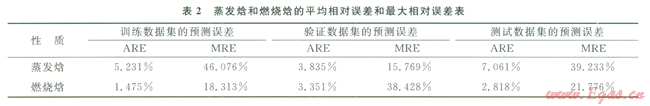
表2中展示了新構(gòu)建燃燒焓和蒸發(fā)焓模型的結(jié)果。從表2中可以看出,經(jīng)過(guò)上述處理后,所構(gòu)建的ANN模型在“驗(yàn)證”集和“測(cè)試”集中的預(yù)測(cè)表現(xiàn)有所改善。
對(duì)燃燒焓而言,盡管“訓(xùn)練”集與另兩個(gè)集之間的一致性不會(huì)直接改善預(yù)測(cè)表現(xiàn),但卻表明了模型的預(yù)測(cè)質(zhì)量有所改善。
同時(shí)還應(yīng)指出,所構(gòu)建的上述模型均重新訓(xùn)練過(guò)若干次,這是因?yàn)?span lang="EN-US">3個(gè)數(shù)據(jù)集是隨機(jī)劃分的。如果用于當(dāng)前訓(xùn)練的數(shù)據(jù)有所不同,最終的ANN模型也會(huì)隨之發(fā)生變化。所以對(duì)有待構(gòu)建的ANN模型的處理過(guò)程并不穩(wěn)定,即可能生成相對(duì)較好或較差的結(jié)果。由此產(chǎn)生的問(wèn)題是,一個(gè)在“測(cè)試”集中表現(xiàn)良好的模型,卻可能無(wú)法以相同的水準(zhǔn)對(duì)其他未包含在數(shù)據(jù)庫(kù)中的純碳水化合物進(jìn)行預(yù)測(cè)。為了解決這一問(wèn)題并獲得更好的模型評(píng)估結(jié)果,我們建議為ANN模型建立一個(gè)共識(shí)方案,以便使用交叉驗(yàn)證的概念。換而言之,就是建立一個(gè)預(yù)測(cè)相同性質(zhì)的模型系統(tǒng),計(jì)算該系統(tǒng)中不同模型的一致性,從而推導(dǎo)出預(yù)測(cè)結(jié)果的準(zhǔn)確性[20]。筆者通過(guò)MATLAB對(duì)此類ANN模型系統(tǒng)的標(biāo)準(zhǔn)沸點(diǎn)進(jìn)行了演示,其結(jié)果展示于圖10中。

從圖l0可以看出,所構(gòu)建的標(biāo)準(zhǔn)沸點(diǎn)模型系統(tǒng)表現(xiàn)穩(wěn)定,有望給出相對(duì)準(zhǔn)確的純碳水化合物預(yù)測(cè)結(jié)果。
3 結(jié)論
此項(xiàng)研究展示了用于計(jì)算和預(yù)測(cè)物理性質(zhì)的各個(gè)模型,而這些物理性質(zhì)則可以用來(lái)開(kāi)發(fā)新型純碳水化合物燃料。所構(gòu)建的模型結(jié)合了QSPR和兩層前饋ANN。建模工作需要被研究純碳水化合物的相應(yīng)分子描述符值和實(shí)驗(yàn)性質(zhì)值。筆者用dragonX[12]軟件計(jì)算了分子描述符值,并從DIPPR801[11]數(shù)據(jù)庫(kù)中獲得了實(shí)驗(yàn)性質(zhì)值,由此構(gòu)建了多個(gè)全面而可靠的模型來(lái)預(yù)測(cè)各種物理性質(zhì),其中包括標(biāo)準(zhǔn)沸點(diǎn)、閃點(diǎn)、燃燒焓、蒸發(fā)焓、液體密度、表面張力、液體黏度和熔點(diǎn)等。然而,構(gòu)建的這些模型也還存在一些缺陷,其中的一個(gè)主要問(wèn)題就是“訓(xùn)練”集和其他兩個(gè)數(shù)據(jù)集之間相對(duì)誤差的不一致性;而另一個(gè)問(wèn)題則是存在會(huì)影響模型預(yù)測(cè)質(zhì)量的不確定性。為了提高一致性,筆者引入了主成分分析(PCA) [8],以進(jìn)一步消除分子描述符值的維數(shù)。實(shí)施后所獲結(jié)果表明:一致性確實(shí)有所提高。為了減少不確定性的影響,建議通過(guò)共識(shí)建模進(jìn)行交叉驗(yàn)證[20],這一思路或許會(huì)對(duì)此有所幫助。所有上述策略均有助于改善所構(gòu)建模型的預(yù)測(cè)精度和質(zhì)量。不過(guò),由于構(gòu)建ANN QSPR模型時(shí)所包含的碳水化合物數(shù)日仍相對(duì)較少,因此所構(gòu)建的這些模型可能還不是很全面,所以應(yīng)開(kāi)展將更多碳水化合物包含在內(nèi)的相關(guān)研究。
參考文獻(xiàn)
[1]GHARAGHEIZI F,ESLAMIMANESH A,MOHAMMADI A H,et al.Determination of critical properties and acentric factors of pure compounds using the artificial neural network group contribution algorithm[J].Journal of Chemical& Engineering Data,2011,56(5):2460-2476.
[2]POLING B E,PRAUSNITZ J M,O’CONNELL J P.Properties of gases and liquids[M].5th Edition,New York:McGraw-Hill,2001.
[3]S0LA D,FERRI A,BANCHERO M,et al.QSPR prediction of N-boiling point and critical properties of organic compounds and comparsion with a group contribution method[J].Fluid Phse Equilibria,2008,263(1):33-42.
[4]KATRITZKY A R,KUANAR M,SLAVOV S,et al.Quantitative correlation of physical and chemical properties with chemical structre:Utility for predietion[J].Chemical Reviews,2010,110(10):5714-5789.
[5]FAULON J L,BENDER A,GOLBRAIK H A.Handbook of chemoinformatics algorithms[M]. London:Chapman& Hall/CRC Press,Taylor&Francis Group.2010.
[6]GODAVARTHY S S,ROBINSON R L Jr,GASEM K A M.Improved structure-property relationship models for prediction of critical properties[J].Fluid Phase Equilibria,2008,264(1/2):122-136.
[7]TODESCHINI R,CONSONNI V.Handbook of molecular descriptors[M].Weinheim(Germany):Wiley-Vch,2002.
[8]ERIKSSON L,JOHANSSON E,KETTANEH WN,et al.Multi and megavariate data analysis:part I-basic principles and applications[M].Umea(Sweden):Umetrics Academy,2006.
[9]AGRAFIOTIS D K,CEDE O W,LOBANOV V S.On the use of neural network ensembles in QSAR and QSPR[J].Journal of Chemical Information and Computer Sciences,2002,42(4):903-911.
[10]GHARAGHEIZI F,SATTARI M.Prediction of triplepoint temperature of pure components using their chemical structures[J].Industrial and Engineering Chemistry Research,2010,49(2):929-932.
[11]ROWLEY R L,WILDING W V,OSCARSON J L,et al.DIPPR 801 property databse,Software Package[G].New York:Design Institute for Physical Property Data,American Institute of Chemical Engineers,2009(http://dippr.byu.edu).
[12]ANON.DragonX Version l.4,Software Package[G].Milano(Italy):Taletesrl,2009.
[13]BRAUNER N,SItACHAM M.Considering precision of data in reduction of dimensionality and PCA[J].Computers& Chemical Engineering,2000,24(12):2603-2611.
[14]GHARAGHEIZI F,BABAIE O,SATTARI M.Prediction of vaporization enthalpy of pure compounds using a group contribution-based method[J].Industrial and Engineering Chemistry Research,2011,50(10):6503-6507.
[15]HAGAN M T,DEMUTH H B,BEALE M.NeuraI Network Design[M].Andover(Massachusetts):International Thomson,2002.
[16]GHARAGHEIZI,F.QSPR studies for solubility parameter by means of genetic algorithm based multivariate linear regression and generalized neural network[J].QSAR& Combinatorial Science,2008,27(2):l65-170.
[17]LERA G,PINZOLAS M.Neighborhood based Levenberg Marquardt algorithm for neural network training[J].IEEE Transactions on Neural Networks,2002,l3(5):1200 -203.
[18]KALOGIROU S A.Artificial neural networks in renewable energy systems applications:A review[J].Renewable and Sustainable Energy Reviews,2001,5(4):373-401.
[19]SUZUKI T,OHTAGUCHI K,KOIDE K.Computer-assisted approach to develop a new prediction method of liquid viscosity or organic compounds[J].Computers& Chemical Engineering,1996,20(2):161-l73.
[20]HANSEN L K,SALAMON P.Neural network ensembles[J].IEEE Transactions on Pattern Analysis and Machine Intelligence,1990,12(10):993-1001.
本文作者:朱子懿 趙興元
作者單位:美國(guó)卡內(nèi)基·梅隆大學(xué)
中國(guó)石油集團(tuán)工程設(shè)計(jì)有限責(zé)任公司西南分公司
您可以選擇一種方式贊助本站
賬贊助")
支付寶轉(zhuǎn)賬贊助
賬贊助")
微信轉(zhuǎn)賬贊助
